
The error of the core-valence approximation for describing core-excited states
Recently spectroscopic methods in the X-ray regime for the study of the electronic and atomic structure of molecules, have seen key experimental advances. Modelling the involved core-excitation processes accurately is, however, rather demanding. One of the reasons is that the core-excited eigenstates are usually encountered in the same energy range as continuum-like valence-excited states. This prohibits bottom-up solutions, where one numerically computes all excited states up to and including the core-excitations, for all but the smallest systems. To overcome this challenge in practice a standard approach is to employ the so-called core-valence separation (CVS) approximation, in which the weak electrostatic coupling between core- and valence-orbitals is neglected. This allows to decouple core-excitation completely from valence-excitations, such that one may solve for the former without interference of the latter. Amongst other codes the CVS approximation is available in adcc, a framework I designed about a year ago in collaboration with the Dreuw group at Heidelberg University. The aim of adcc is to facilitate the development of novel excited-states methods based on the algebraic-diagrammatic construction scheme for the polarisation propagator (ADC) and the code has already been involved in a number of recent publications, for example to simulate electronic spectra in the UV/vis and the X-ray regime.
 |
||
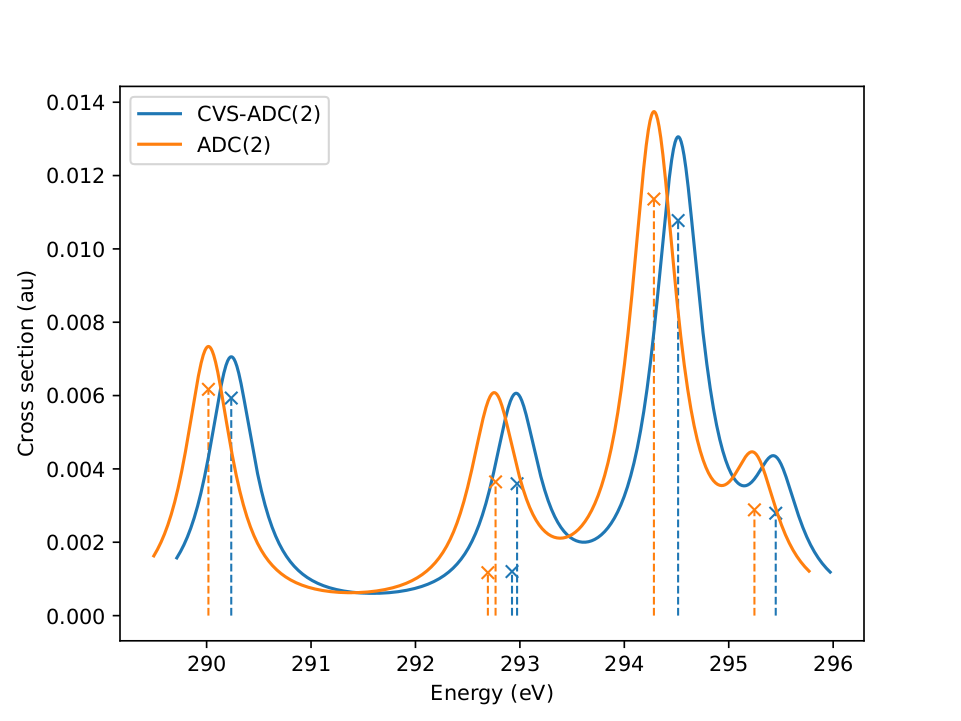
| Comparison of core-excitation spectrum of the carbon edge of 1,2-difluoroethane as obtained at approximated CVS-ADC(2) and full ADC(2) level. Our recent study on the CVS error confirms for the K-edge and a representative set of main-group compounds that the CVS approximation conserves the good description of spectral features of the underlying excited-states method, here ADC(2), while being numerically more feasible. |
In collaboration with Thomas Fransson I have recently been able to construct an refinement scheme, the CVS relaxation, which is able to remove the error from a CVS-approximated solution. We implemented this scheme using the adcc code and used it to characterise the CVS error systematically over a large range of realistic molecules and basis sets for the ADC family of methods, see our publication. The takeaway is that for K-edge spectra (where the 1s electrons are excited) the CVS error is small (around 0.5 eV) and only gives rise to a shift of the simulated spectrum, see the figure above. This shift is moreover roughly constant across different molecular systems as long as the same element is probed, but shows a dependence on the employed basis set, where it is especially small for basis sets providing a balanced description of core and valence regions. In ongoing work Thomas we want to extend our study to other X-ray spectroscopies (e.g. L-edge).
Highlighted publications
- Michael F. Herbst and Thomas Fransson. Quantifying the error of the core-valence separation approximation. Journal of Chemical Physics, 153, 054114 (2020). Part of JCP Emerging Investigators, received Editor's Pick. DOI 10.1063/5.0013538 [code] Blog article.
- Michael F. Herbst, Maximilian Scheurer, Thomas Fransson, Dirk R. Rehn and Andreas Dreuw. adcc: A versatile toolkit for rapid development of algebraic-diagrammatic construction methods. WIREs Computational Molecular Science, 10, e1462 (2020). DOI 10.1002/wcms.1462 [code] Blog article.