The development and understanding of new materials play a key role for major
industrial and societal challenges of the 21st century.
Over the past years more and more materials have been design fully in silico.
At the core of this are automated workflows,
which systematically perform calculations on thousands to million of compounds.
Compared to the early years of materials modelling,
which was centred around performing
a small number of computations on hand-picked systems,
such high-throughput screening approaches have much stronger requirements
with respect to the robustness of employed algorithms:
even a small percentage of erroneous or failing calculations equals
a large number of cases, which need to be checked by a human (causing idle time)
and redone thereafter (causing a noteworthy computational overhead).
In my research I want to overcome these obstacles
and accelerate computational materials discovery by
providing more reliable materials simulations
featuring robust error control
and built-in uncertainty quantification.
Achieving these aspects will not only contribute crucially to reducing
the human time required to setup and verify calculations
(the typical bottleneck in high-throughput workflows),
but it will also enable promising prospects
such as active learning or adaptive numerical schemes,
which automatically balance errors in order to
obtain a simulation result along a path of least computational effort.
This work is inherently a multidisciplinary research effort
and in my work I frequently collaborate across domain boundaries.
A central component of my efforts is therefore to develop interdisciplinary software platforms
which allow all involved communities to synergically join their forces.
Building on top of my codes I explore emerging opportunities
such as mixed-precision linear algebra
or algorithmic differentiaton
for materials modelling and investigate their perspectives to obtain
more efficient simulations or to better integrate physical models
with data-driven approaches.
-
My collaborations span across the disciplines and include
application scientists, mathematicians and computer scientists alike.
Recently I have joint multiple interdisciplinary research initiatives
in which I take a mediating role between the involved communities,
for example by providing interdisciplinary software platforms,
such as the Density-Functional ToolKit (DFTK),
in which our …
read more
-
Working side-by-side with mathematicians and application scientists
(see also collaborations) on a daily basis
has allowed me to learn how to mediate and lower barriers between the disciplines.
Apart from problems in the terminologies used in the individual subjects
a frequent issue is the lack of software, which allows multiple …
read more
-
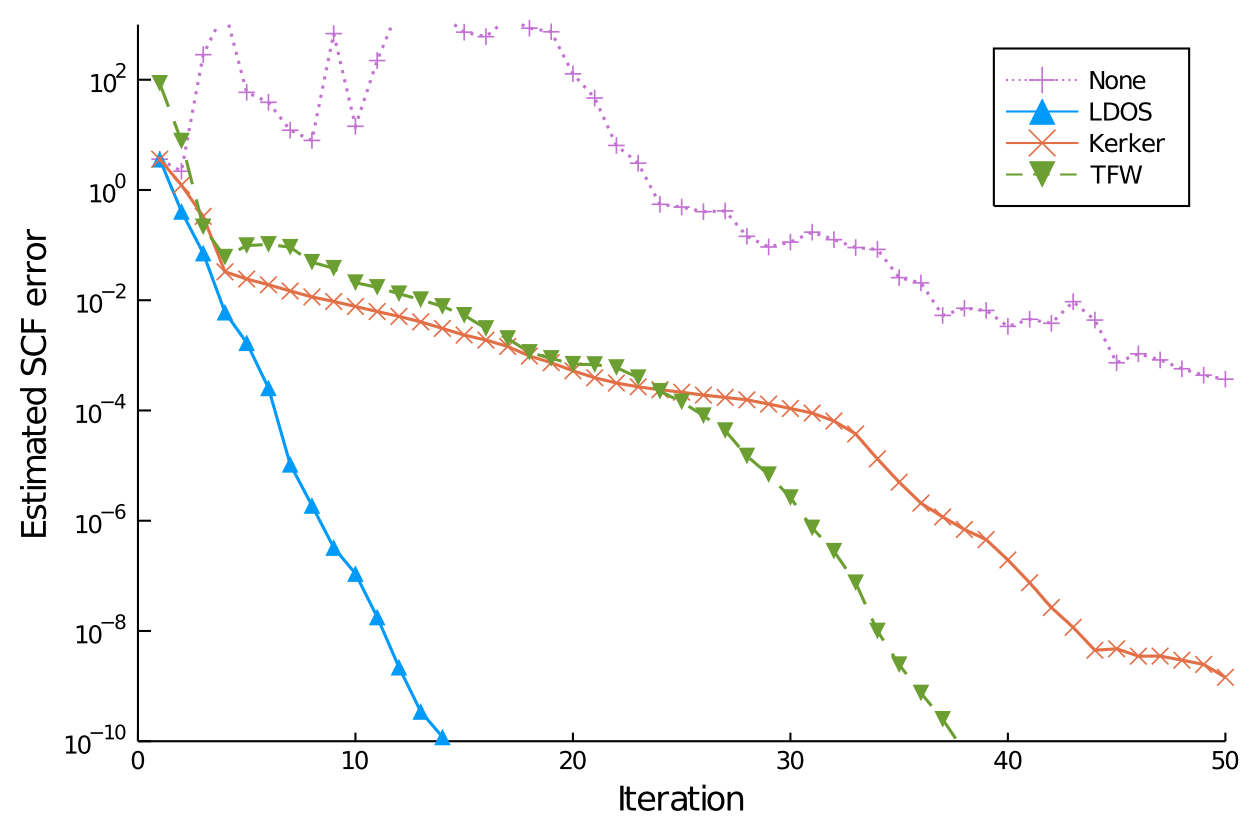
In state-of-the-art DFT algorithms a sizeable number of computational parameters,
such as damping, mixing/preconditioning strategy or acceleration methods,
need to be chosen. This is usually done empirically, which bears the risk
to choose parameters too conservatively leading to sub-optimal performance
or too optimistically leading to limited reliability for challenging …
read more
-
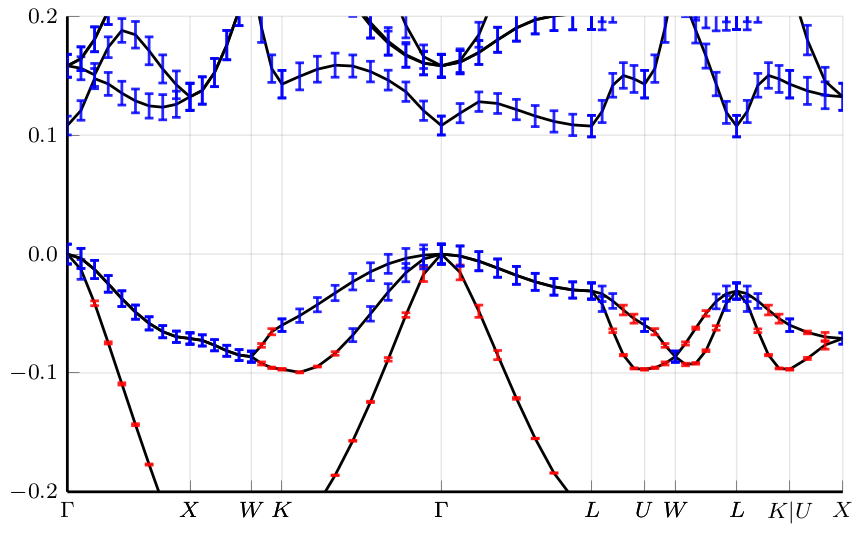
Due to the involved multi-scale nature of standard materials simulation workflows
rigorous techniques for error estimates or uncertainty quantification (UQ)
are underdeveloped and error analysis on simulation results is rarely attempted.
However, recent activities to equip data-driven interatomic potentials
with built-in uncertainty measures as well as recent progress
on a …
read more
-
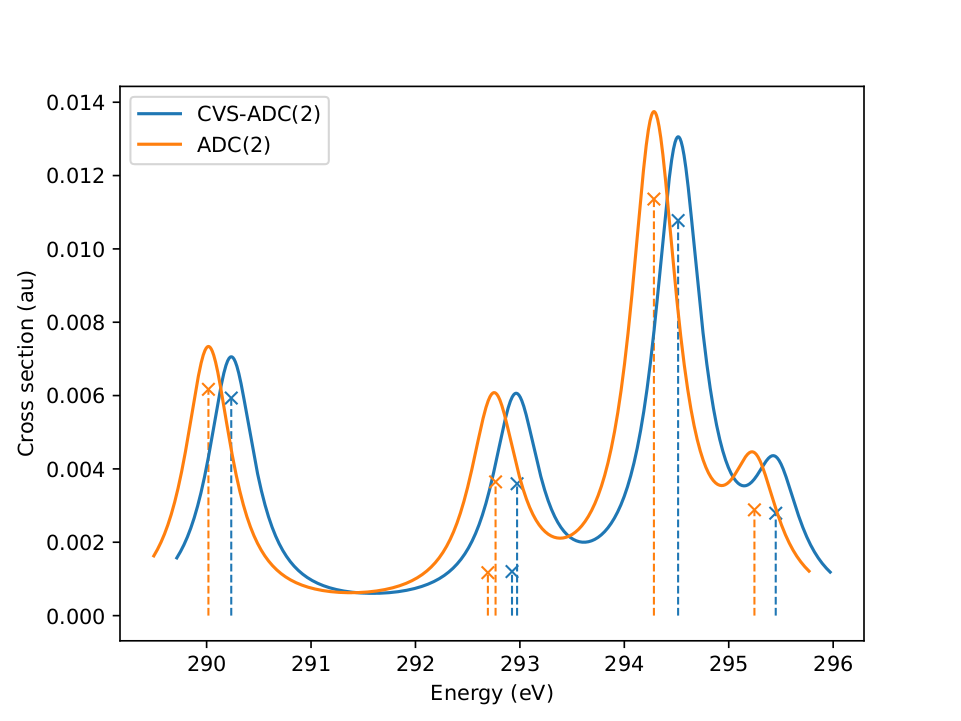
Recently spectroscopic methods in the X-ray regime
for the study of the electronic and atomic structure of molecules,
have seen key experimental advances.
Modelling the involved core-excitation processes accurately is,
however, rather demanding.
One of the reasons is that
the core-excited eigenstates are usually encountered in the same energy
range …
read more
-
In the first half of my PhD studies in the Dreuw group in Heidelberg
I investigated novel approaches
for solving the Hartree-Fock (HF) problem using the finite-element method (FEM).
The FEM is a standard approach for solving partial differential equations
and the aim of the project was to apply it …
read more
-
In the second half of my PhD studies
I turned my focus towards developing
quantum-chemical simulation methods
using Coulomb-Sturmian basis functions.
Coulomb Sturmians are atom-centred, exponentially decaying functions,
which show some desirable properties.
For example they are complete and computing the required
two-electron integrals is simpler compared to Slater-type orbitals …
read more